Son Zamanlarda Adını Sıkça Duyduğumuz SMA Tam Olarak Nedir?

1) Nedir, ne değildir?
uzunca bir süredir sosyal medya üzerinden yapılan sma'lı ayşe bebeği kurtaralım minvalindeki yayınlardaki artış üzerine bir sağlık çalışanı olarak paylaşma gereğini duydum. yazı uzunca oldu, umarım tümünü okuma şansınız olur, kusuruma bakmayın şimdiden..
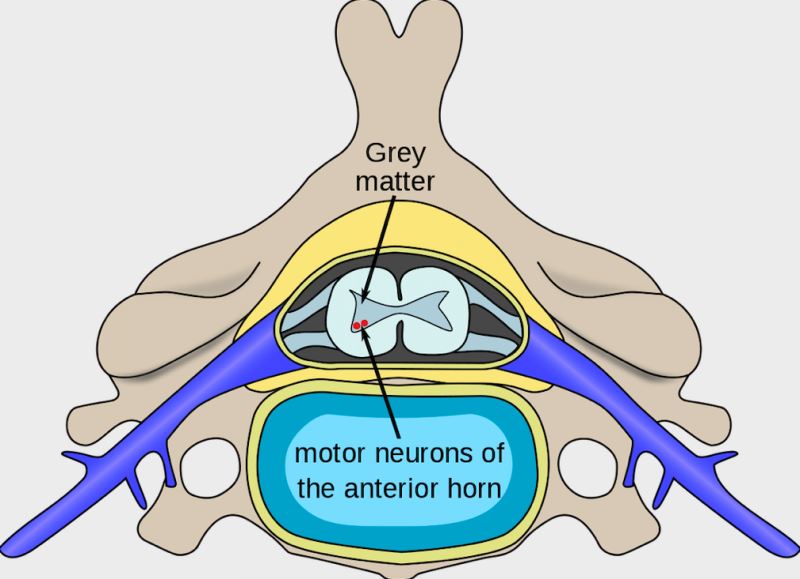
SMA (Spinal muscular atrophy): Omurilikte bulunan ön boynuz motor sinir hücrelerini etkileyerek hareket kabiliyetini kısıtlayan bir kas hastalığıdır.
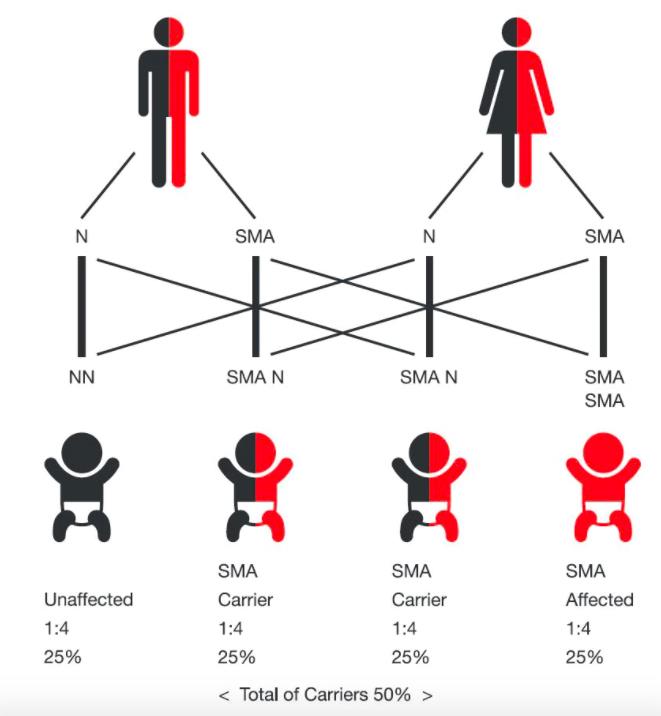
nadir hastalıklar %80’i genetik kaynaklı olan, genel nüfusun yaklaşık %6-8'ini etkileyen kronik, ilerleyici bozukluklardır. sorun, çoğalma hücresi oluşumunda nedeni belirli olmayan bir gen kusuru ortaya çıkması ve bunun da son tahlilde yumurta/sperm üzerinden temel hücreye, oradan da embriyoya ve bebeğe nakledilmesi durumudur. bu kusurlu gen nereye aitse ilişkili olduğu metabolizma sistemini de kusurlu hale getirir, çok kabaca; hatalı bir yazılım kodunun tüm yazılımı çökertmesi durumuna benzetebiliriz. bu genetik kusur; hemen tümümüzde var, pek çoğunu taşıyıcı olarak; belirti göstermeden içimizde bulunduruyoruz.
yine kabaca; kusurun yavruda belirmesi için benzer kusurun bir başka hücreyle birleşerek (anne/baba cinsiyet hücreleri birden) aktifleşmesiyle ortaya çıkıyor. yani siz veya eşiniz hangi kusurlu geni taşıdığınızı asla bilemeyebilirsiniz. aynı gen kusuru her ikinizde de varsa… orada da görülme ihtimali %100 değil, yine kabaca her gebelik için % 25 gibi.. ve bu sadece insanda değil, tüm memelilerde söz konusu… yani bu kötü bir piyango ve hepimizde ortaya çıkabiliyor. sıklığı; her 100.000 kişide 40 hasta yaklaşık olarak.
en bilinenlerini yazayım buraya – talasemi –orak hücreli anemi-ailesel akdeniz ateşi-osteogenesis imperfekta (cam çocuk), ms (multipl skleroz), kistik fibroz, als (suna kıraç-hawking görülen hastalık) vs. bir de bunların ultra-nadir tipleri var; 100.000 kişide 2 hasta… küresel olarak dünyada 64 milyon bireyin ultra nadir hastalığı olduğu öngörülüyor; ülkemizde ise akraba evliliklerinin (tüm evliliklerin yaklaşık %20’si) çok sık (aynı genetik kusurun hala kızı/amca oğlunda var olup katmerlenmesi) ve ailelerin çok çocuklu olması (her doğumda %25 kusurlu bebek- 4 çocuğun dördünde birden olma ihtimali) nedeniyle yaklaşık 30.000 civarında ultra-nadir hastalığı bulunan birey görülüyor. yüksek sayı bir felaket, fakat daha da kötüsü bu hastalık gruplarının sadece %5’i tedavi edilebiliyor. kalanı, destek tedavileriyle hayata tutunmaya çalışıyor. nadir hastalıkların çoğunun etkin bir ilacı olmadığı gibi bu hastalıklar için kişiye özel, yenilikçi ilaçlar geliştirilmesi çok pahalı, uzun ve zorlu bir süreci içeriyor.
covid'den hatırlayalım; ilaç geliştirme, toksikolojik ve preklinik hayvan çalışmalarından başlayarak ilacın insanlardaki etkinlik ve güvenilirliğini değerlendirmek amacıyla sağlıklı insanda ve geniş hasta gruplarında yapılan çoklu faz klinik araştırmalardan sonra gerçekleşmekte. şimdi bunları hem çocuklar üzerinde yapacağız, ayrıca sayıları bu kadar düşük olan hasta grupları üzerinde etkinlikleri araştırılacak; zaten o yüzden tedavilerini bulmak çok güç (maalesef %5). ayrıca bozuk genin durumuna göre bir de tip 1-2-3 gibi şiddet grupları ortaya çıkıyor, bu da tedaviyi daha da zora sokuyor. bir kısmı çocuklukta, bir kısmı ise ilerleyen yaşlarda ortaya çıkan bu gen anomalilerinde tanılanmış, klinikleri ve tedavileri (olabildiğince) belirlenmiş 60’a yakın hastalık mevcut; konumuz olan sma’da bunlardan biri, 10.000 canlı doğumda 1 görülür, taşıyıcılık oranı toplumda 50’de 1 (yani her 50 bireyden birinde bu gen bozuk; bir de yakın akraba evliliği). türkiye’de yaklaşık 3.000-3.300 sma hastası (tip 1, tip 2 ve tip 3 dahil) olduğu ve her yıl 180- 200 kadar ağır formda yeni sma vakasının eklendiği tahmin edilmekte.
tüm bu güçlüklere rağmen, son birkaç yıl içinde küçük moleküller, antikorlar, rna ve gen tedavisine dayalı kişiye özgü ilaçların geliştirilmesi (kişiselleştirilmiş tıp); ayrıca teknolojik yenilikler sayesinde, en ciddi genetik hastalıkların tedavisi için bile umut vadeden sonuçlar sunulmakta. ancak bu tedaviler oldukça maliyetli. üstelik bu ilaçlarla her vakada tedavi (tip 1-2-3 vs farklılıkları) sağlanabileceği de söylenemez. tedavinin başarısını etkileyebilecek çeşitli faktörler var.
sma’yı sık sık duyuyoruz, ancak kistik fibrozdan, cam çocuğa kadar diğerlerini neden bu kadar duymuyoruz?
çünkü bunlara has ilaçlar ya yok; ya var olan kimi “idare ettirici kortizon gibi” ilaçlar ucuz, ya dernekleri yok ya da ve en kötüsü ardında novartis gibi, gen ilaç gibi firmalar, ilaç tröstleri yok.. bu devlerin sosyal medyayı ne kadar iyi manipüle ettiklerini, ürünlerini satmak için kimleri nasıl kullandıklarını, gereğinde hastaları bile bu işe alet ettiklerini çok gördüm, hiç unutmadım. bundan dolayı sosyal medya ‘da flood’ladıklarına, bot mesajları “up” ladıklarına yürekten inanıyorum.
firmaların bu yaklaşımları nedeniyle de; hastalara ilaç temini konusunda geri ödeme kurumları mecburen rasyonel davranmak durumunda kalıyor (bu genel yaklaşım- ülke bazında düşünülmemesini rica ederim). çözümcül yaklaşımda hasta örgütleri vasıtasıyla nadir görülen hastalıklar hakkında toplumun farkındalığının artırılması çabaları (ayşe bebeğe ilaç gelsin şeklinde değil, her sene yeni 200 hastaya fayda böyle sağlanamaz) dünya çapında ilaç gelişimine öncülük eden ‘yetim ilaç yasası'nın oluşturulması ve klinik araştırmalar için hasta bulunmasına katkılar (yasal engellerin gevşetilmesi) yadsınamaz. örneğin sma için madem akraba evliliklerini engelleyemiyoruz en azından bir yenidoğan tarama programı içine alarak hastalık etmeni smn1 geninin bu tür evlilik yapanlarda taranması başlatılabilir (bu pahalı bir gen arama tekniği ama inanın ilaçtan çok daha ucuz), tedavi edici değil, koruyucu hekimlik adına katkılar sağlanabilir. ilaç tröstleri üzerine stk baskıları ile ürünlerin fiyatları aşağıya çektirilebilir. böylece kısıtlı sağlık kaynaklarımız sadece sma için değil, yukarda sayılan ve sesleri de pek çıkmayan çocuklarımızla ilgili onlarca –diğer- genetik hastalık tanı ve tedavisinde kullanılabilir.
2) SMA'nın gündelik hayattaki yeri üzerine
sma ile boğuşan çocukların ailelerini vicdansızca eleştiren yazılar görmeye başladım son zamanlarda. ya neden bahsettiklerini sahiden bilmiyor bu insanlar ya da gerçekten bir takım erdemlerden yoksunlar. açıklayalım...
sma için bulunan tedavi çok çok yeni. aradan çok vakit geçtiği için bazı şeyleri yanlış ya da eksik hatırlıyor olabilirim ama genel hatlarıyla olay şöyle gelişiyor:
2011 senesinde, amerikalı bir çiftin avery canahuati adını verdikleri bir kızları oluyor. avery'de fark ettikleri bazı sorunlardan ötürü doktora gidiyorlar ve bebeğe sma tip 1 tanısı konuluyor. o dönemde, sma hastası bebeklere bir şans sunabilecek herhangi bir tedavi henüz yok. ailesine, kızlarının iki yaşını göremeden öleceği söyleniyor.
bunun üstüne avery's bucket list ile aile avery'nin henüz anlayamayacağı ama en azından kendilerine saklayabilecekleri anılar biriktirmeye başlıyor ve böylece tedavi geliştirilmesi için başlattıkları kampanyanın da ses getirmesini amaçlıyorlar. ailenin bir başka amacı da sma gibi genetik hastalıkların hamileliğin ilk aylarında yapılan taramalara dahil edilmesini sağlamak zira o yıllarda, amerikalı bir aile doğacak çocuklarında olabilecek birçok hastalıktan sadece sma'ye baktırtmak için dahi 2500 dolar para ödemeliymiş. onlarca genetik hastalığın olduğu şu hayatta, bir ailenin her bir test için iyi ihtimalle 2500 dolar para ödemesinin imkanı yok.
hayatımda iki bilimsel araştırmayı düzenli destekledim: biri, rett sendromu gen tedavisi için yürütülen bir çalışmaydı. diğeri, sma gen tedavisi (zolgensma) çalışmasıydı. hedef bir milyon dolardı. yine hatırlamadığım bir sebepten ötürü zolgensma araştırması fonlanmıyordu (ya da aktarılan kaynaklarda ciddi kesintiler vardı) ve ailenin dikkat çekmek istediği nokta buydu. o aile, kızlarının iki yaşını görmeden öleceğini biliyordu. amaçları, başka çocukları kurtarmaktı.
nitekim, gereken para fazlasıyla toplandı, gereken farkındalık yaratıldı, sophia's cure foundation aracılığıyla parayı gerekli yerlere ulaştırdılar ve muhtemel bir gen tedavisi için yürütülen çalışmalara yeniden başlandı (ya da hızlandırıldı).
aynı zamanda, sma ile beraber nadir görülen 20 genetik hastalığın araştırmalarının fonlanması için çıkmasını istedikleri yasa tasarısının da önünü açtılar ve üstelik sma, gebelik taramalarına dahil edildi.
zolgensma'nın bulunmasındaki başarı bana kalırsa çok büyük oranda canahuati ailesinin başarısıdır. benzer bloglar, benzer hikayeler, benzer kampanyalar olmasına rağmen en büyük etkiyi yaratanlardan biriydi bu aile. yazının başında "olaylar gelişiyor" dememin sebebi buydu. ben, zolgensma'nin fda onayı aldığı günü hala hatırlıyorum ve parçası olmaktan en çok gurur duyduğum şeylerden birisidir bu.
avery 5,5 aylıkken öldü. ailenin, avery'den sonra ivf ile başka bebekleri oldu. böylece doğan çocuklarının sma-free olduklarını biliyorlar.

avery'nin hikayesi böyleydi, geleyim olayın diğer yüzüne
ailenizde sma tanısı almış birinin olmaması sizin taşıyıcı olmadığınız anlamına gelmiyor. daha öncesinde sağlıklı bir ya da birkaç çocuk sahibi olmuş olmanız da ikinci, üçüncü, yedinci çocuğunuzun sma hastası olmayacağı anlamına gelmiyor. sma, otozomal resesif geçişli bir hastalık. yani, siz ve partneriniz hayatınızı taşıyıcı olduğunuzdan haberiniz dahi olmadan sorunsuz bir şekilde sürdürebilirsiniz ama hasta bir çocuğunuz da olabilir. çocuklarınız hasta olmasa dahi taşıyıcı olabilirler ve sonraki kuşaklar da hastalık riskini taşımaya ve aktarmaya devam ederler. sma-free çocuk sahibi olacağınızı bilmenizin tek yolu genetik tarama ve taşıyıcı olduğunuzu biliyorsanız da ivf. bunu da bireysel olarak herkesin yapması hem maddi açıdan mümkün değil, hem de bu bilinci oluşturmak çok zor.
hasta bir çocuğu olduktan sonra zeka gerisi bir şekilde ikinci çocuğu herhangi bir önlem gözetmeden yapan zırcahil birisi varsa yazın sosyal medyaya, beraber gömelim ama bu ailelerin sma'lı çocukları gördüğüm kadarıyla ya ilk çocukları ya da ilk hasta çocukları. yakın çevrelerinde sma hikayesi de yoksa şayet, bu insanların, bebeklerinin ölümcül bir hastalıkla doğacağını düşünmeleri için hiçbir sebepleri yok.
sosyal medyaya bakıyorum da çocukların ailelerine bol keseden atanlar var. benim sma hastası tanıdığım kimse yok mesela. taşıyıcı olmadığımı da biliyorum ama bu tip hastalıklar için genetik taramaların yapılmadığı bir ülkede yaşıyorsanız bireysel çabanızla kaç hastalığa baktırabilirsiniz böyle? kafanızı kullanın biraz. sma riskinin bittiği noktada xeroderma pigmentosum başlar, o biter duchenne muscular dystrophy başlar, o biter tay sachs başlar, o biter başkası başlar.
bir çocuğun tedavisi için 2,5 milyon dolar para harcanır mı? harcanır elbet. ama bu sayı her yıl 200 çocuk olunca işin rengi değişir.
hızla yapılması gereken ilk şey, canahuati ailesi gibi birçok ailenin abd'de verdikleri savaş sonucu aldıkları verimli sonuca da bakarak türkiye'de de sma gibi nadir görülen genetik hastalıkların bir an önce gebelik taramalarında ücretsiz ve zorunlu olarak uygulanmasını sağlamak olmalı. nadir görülen genetik hastalıkların kaçı taranıyor, ne kadar erişilebilir oluyor bu taramalar bilgim yok ama şu anki duruma bakarak en azından sma'nın bu taramalara dahil olmadığını anlayabiliyorum.
devletin, sma tedavisini karşılaması için değil, ilk olarak bu hastalıkla doğacak çocukların önlenebilmesi için çalışılmalı. sma önlenebilir bir hastalık ama bireysel olarak herkesin bunu karşılayabilmesi imkansız.
1/6000 sıklıkla ortaya çıkıyor sma. iyi ihtimalle, senede 200 çocuğun sma hastası olarak doğduğu ülkede, her bir çocuğun tedavi masrafına bu kadar para harcamak devletlerin gözden çıkarabileceği bir miktar değil. zaten gözden çıkarması mantıksız olur. geri kalan her yıl yeni doğan 1.1 milyon bebeğin ve milyonlarca çocuğun sağlıklı tutulması ve sağlık sistemine ulaşabilir olmaları gerekiyor. devletin kaynakları sonsuz değil, bahsedilen miktarlarsa muazzam.
bilmem kimi yaşatalım gibi kampanyalara maddi destek vermiyorum. sorunun çözümünü bireysel kampanyalarda görmüyorum. yapabileceğim bağışları da zamanında yaptığım gibi tedavi geliştirme odaklı bilimsel projelere aktarmayı kendi adıma daha etik ve daha çözüm odaklı buluyorum. sma tedavisinin geliştirilmesinde mini minicik de olsa bir katkım olmuş olması beni haklı çıkarıyor.
burada önemli iki nokta var bana kalırsa: öncelikle, yukarıda bahsettiğim gibi nadir görülen genetik hastalıklar için gerekli olan taramaların yapılmıyor oluşu; ikinci olarak da çözümü bulunmuş bir hastalıkta ilaca ulaşılamıyor olunuşu. bu iki durum da kabul edilebilir durumlar değiller.
semptom göstermeye başlamadan önce spinraza tedavi almaya başlayan çocuklar, şurada görülebileceği üzere düzenli tedavi ile gayet normal bir hayat yaşayabiliyorlar:
Duygu Özaslan'ın da Hastalığı Olan Bulimia Nervoza Nedir?



